第3話「ショ糖密度勾配遠心法」
籔井教授 今日は、超遠心機を使ったウイルスの精製の中核となる密度勾配遠心法の話をします。原理的には、速度ゾーン遠心法 (rate zonal centrifugation)と等密度遠心法 (isopycnic centrifugation)とに分けられます。
① 速度ゾーン遠心法 (rate zonal centrifugation)
② 等密度遠心法 (isopycnic centrifugation)
③ 平衡密度勾配遠心法 (equilibrium density gradient centrifugation)
ウイルスの精製で、最も使用頻度が高いのが速度ゾーン遠心法です。最初にこの遠心法についてお話しますが、たぶん今日はこの話だけで終わるでしょう。
ウイルスを速度ゾーン遠心法で精製するには、ほとんどの場合、ショ糖(スクロース)溶液の密度勾配を使うので、現実的にはショ糖密度勾配遠心法 (sucrose density gradient centrifugation) と同義語のようなものです。粒子の大きさと形、つまり前回お話ししたS値(沈降速度)の異なる粒子が異なる速度で沈降していくので、帯状のゾーンを形成します(下図)。それで速度ゾーン遠心法というわけです。

早斐 第2話で教授がお話しくださった分画遠心法でも、S値の異なる粒子が異なる速度で沈降していくからこそ、一定の回転数、つまり一定の相対遠心力の下では、遠心時間によってペレットに来る粒子がS値の大きいものからだんだん小さいものになっていきますよね。どこが違うんですか?
籔井教授 S値の差を利用して分けるということから言えば、あなたの言う通り分画遠心法と原理は同じです。ただ、単一濃度の溶液ではなく、ショ糖溶液の密度勾配、例えば濃度が10%(w/v)から60%(w/v)の密度勾配の中を沈降させることによって対流が解消されるとともに沈降がゆっくりになるので、異なるS値に応じたバンド(速度ゾーン)が形成されやすくなるのではないかな。
早斐 そうすると、私たちがよくやるショ糖クッション遠心 (sucrose cushion centrifugation) は、分画遠心法と速度ゾーン遠心法との中間的位置づけでしょうか?
籔井教授 今日は出だしからずいぶん突っ込んできますね。
ショ糖クッション遠心法は、通常、ショ糖密度勾配遠心法の変法とみなされていますが、そのように考えてもいいと思います。密度勾配遠心法で精製をやっていく上で、ちょっと困るのが密度勾配の上に乗せられるサンプル溶液の量が少ないことです。おおざっぱに言えば、密度勾配溶液の10分の一が目安です。
密度勾配に重層するサンプルの量は、密度勾配溶液の容量の10分の一が目安です。
例えば、スウィングロータ SW 32 Tiには遠心チューブ1本あたり33 mL(公称容量は38 mL)乗せられますから、ショ糖密度勾配溶液は30 mLにして3.0 mLのサンプル溶液を重層するという意味です。あくまでも目安ですから、4 ~5 mLくらい入れてもいいのですが、多ければ多いほどバンドがブロードになり、分離が悪くなります。
早斐 この前のステップの分画遠心、つまり最初の超遠心でのペレッティングでは、固定角ロータType 45 Tiを使いました。それでPCボトルキャップアセンブリの底にできたペレットを、濃縮しすぎはNGと言われて1本あたり5 mLに懸濁しましたから、30 mLもの処理すべきサンプル溶液があります。30 mLを6本に分けると、1本あたり5 mL。5 mLを重層するのはやめたほうがいいんですね。でも、1本あたり3.3 mLとすると9本になるので、ショ糖密度勾配遠心のステップにスウィングロータ SW 32 Tiを使っても1回の遠心では終わらないわね。
籔井教授 あまり固定的に考えないでください。
1本あたりどれくらいロードするかは、①遠心を何回やるか、②次のステップの遠心があるのか(これが最後の精製ステップなのか)、③収量を優先するか、純度を優先するか、などを勘案して、作業が最も効率的になるように決めればよいのです。
ウイルスの精製にあたって、1本あたりどれくらいロードするかを決めるヒント:
① ロータの大きさと同じ遠心を何回やるか
② 次の精製ステップにも遠心があるのか(これが最後の精製ステップなのか)
③ 純度と収量のどちらを優先するか
早斐 収量か純度か、ですか。それは精製したウイルス粒子を何に使うのか、その目的を考えて精製しなさい、ということですね。
籔井教授 例えば、ロタウイルス粒子の物理化学的性状の研究を目的として、完全粒子(triple-layered particle: TLP)と2層粒子(double-layered particle: DLP)を分離してその違いを比較するとなると、収量を犠牲にした究極的なレベルの精製が要求されるでしょう。一方、ロタウイルスのゲノムRNAだけがたくさん欲しいのであれば、核酸を含んだ他の粒子や遊離の核酸のコンタミネーションがなければいいわけです。
早斐 私の場合、当面は後者だわ。次世代シーケンシングをやるので、余計な核酸がなくロタウイルスのゲノムRNAが濃縮されている方が、その後の解析にずっと効率的ですから。
籔井教授 別の言い方をすれば、目的のウイルスが100%ある分画に来て、他の夾雑粒子は0%という「うまい話」はない。教科書に出てくる「絵の世界」のような固定的イメージを持ってはいけません。
早斐 「絵の世界」というのは、教授が最初に描いたショ糖密度勾配法の概念を示す絵のことですね。
籔井教授 そのとおり。絵は初心者の理解を助けるためのものです。絵は現実を理想化していることが多いのです。プロはそこから抜け出さなければなりません。
さて、ポイントは単純な分画遠心、つまりペレッティングでも出発材料の100%がペレットに来るわけではなく、逆に100%を目指せば本来除きたかった夾雑粒子が紛れ込んでくるということです。
早斐 ウイルスはもれなく回収したいけれども、それより微小な夾雑粒子のクロス・コンタミネーションはいやですね。
籔井教授 あくまでも目安ですが、ものの本によると、2つのS値の異なる粒子を遠心すると、S値の大きい粒子が完全にペレットに来るときに、S値の小さい粒子がペレットにコンタミする割合は、おおざっぱに言って2つのS値の比に比例するそうです。
さて、ロタウイルスの精製を例にお話しすると、このショ糖密度勾配遠心後、0.8 mL~1.0 mLずつ、33~41に分画してウイルスが存在する画分(フラクション)を同定し、プールすることになります。どこからどこまでのフラクションを回収してプールするかは、収量と純度に大きく影響する因子です。ここでも①精製のステップと②実験の目的を考えて決断しましょう。
早斐 目的とするウイルス、ここではロタウイルスが存在するフラクションをどうやって見つけるのですか?
籔井教授 ウイルス粒子は何でできていますか?
早斐 もちろん、タンパク質と核酸です。ウイルスとは、a piece of bad news wrapped up in a protein (タンパク質の殻に包まれた悪い情報)だと英国の免疫学者であるピーター・メダウォー(1915-1987)が言った、と教授から何度もお聞きしました。
籔井教授 ごめんなさい。齢をとると腰痛が始まり同じ話を何度もするようになるから、ご寛容を。ハンガリー出身の免疫学者でEBウイルスの研究で有名だったジョージ・クライン(1925-2016)が、The stupidest virus is cleverer than the cleverest virologist(一番愚かなウイルスでも一番頭のいいウイルス学者より賢い)と言ったという話はまだしていないかな?
早斐 初耳です。
籔井教授 ジョージ・クラインがどこでそう言ったか知らないけれど、ドロシー・クローフォードというエジンバラ大学の教授が “Viruses: A Very Short Introduction”(2nd ed., Oxford University Press, 2018)という、タイトルからして想像がつく実に薄い本の最後のページに紹介しています。
伊藤 教授、本題からそれないようにお願いします。
籔井教授 早斐さん、タンパク質と核酸を検出するにはどうしますか?
早斐 タンパク質も核酸も紫外部に吸収があります。タンパク質は280 nm、核酸は260 nmに最大吸収があるので、分光光度計で各フラクションの紫外部吸収を測定します。タンパク質はLowry法やBradford法でも測定できます。
籔井教授 よろしい。さらに、各フラクションのショ糖の質量濃度は屈折計で測る屈折率から換算できます。今はロタウイルス粒子の精製をしているわけだから、これを特異的に検出するにはどうしたらいいだろうか?
早斐 ウイルスタンパク質には抗原性がありますから、ELISAで各フラクションの活性を調べます。また核酸の方は、何か特定の遺伝子分節、例えばVP7遺伝子の全域を増幅できるBeg9とEnd9のプライマー・ペアを使ってRT(reverse transcription)-PCRで増幅する、またはNSP3遺伝子を標的としたリアルタイムPCRで検出すれば、半ば定量的な判定もできます。私はやりたくないけれど、各フラクションの一部をMA104細胞に接種してみれば、感染性のある粒子が存在するかどうか分かると思います。これも現実的かどうか分かりませんが、各フラクションの電子顕微鏡観察をすれば、粒子の性状が分かると思います。
籔井教授 ずいぶんスラスラと上げましたね。
ところで、ロタウイルスのゲノムRNAをもっと簡単に直接見る方法はありませんか?
早斐 あっ、ポリアクリルアミドゲル電気泳動法(PAGE)ですね。各フラクションから抽出したRNAを10%のポリアクリルアミドゲルで流せば、RNAの泳動パターンからロタウイルスA(RVA)であることが即座に分かりますね。
籔井教授 大切なのは、それぞれの検出法で何に着目してウイルスを見ているのか、常に頭に入れておくことです。まとめてみましょう。
非特異的検出法:
① タンパク質:紫外部の吸収(280 nm)、タンパク質定量法(Bradford法、Lowry法)
② 核酸:紫外部の吸収(260 nm)
特異的検出法:
① 形態という観点からみると:電子顕微鏡観察
② 抗原性という観点からみると:ELISA
③ ゲノム核酸という観点からみると: RT-PCR、リアルタイムPCR、PAGE
④ 生物活性(感染性)という観点からみると: MA104細胞に接種して細胞変性効果(CPE)
早斐でも、PAGEはRT-PCRやリアルタイムPCRと比べると感度が悪くありませんか?
籔井教授 実は、RT-PCRやリアルタイムPCRは感度が良すぎるのです。事実上、すべてのフラクションがロタウイルスのRNA陽性になってしまいます。目的は、フラクションのどこからどこまでプールすれば精製されたウイルス粒子がたくさんとれるかということですから、PAGEの感度がちょうどいいくらいです。ELISAも感度がいいので、ほとんどのフラクションが陽性になります。しかし、グラフを描けば簡単にピークが分かりますから、フラクションのどこからどこまでプールすればいいか判断できます。もちろん、どこまでプールするかは収量と純度に大きく影響する因子ですので、①精製のステップと②実験の目的を考えて決断しましょう。
伊藤 そろそろ今日の時間も残り少なくなってきましたので、最後にショ糖クッション遠心についてのお話をお願いします。
籔井教授 繰り返しになりますが、オーソドックスなショ糖密度勾配遠心法による精製の弱点として、乗せることのできるサンプルの容量が少ないことがあります。
伊藤 教授は、密度勾配溶液の10分の一を目安に乗せるように、とおっしゃいました。
早斐 ベックマン・コールター社のウェブサイトを見ると、ベックマン博士は4~5%にするようにおっしゃっているみたいですけど、これだともっと大変ね。
籔井教授 誰がどう言っていてどちらが正しいかではなく、何度も言っているように、目的である純度と収量を勘案して決めることです。ただ1つ注意したい初心者の誤りは、乗せられる量が少ないからというので、前段階のペレッティングのところでペレットを少量のバッファーに懸濁してしまうことです。私の経験からすると「10分の一原則」を守ってもらうのが、大きな損失につながらない安全弁になると思います。
さて、この弱点を補い大量のサンプル溶液を乗せられるのがショ糖クッション遠心です。ただ、オーソドックスなショ糖密度勾配遠心法で目的とするウイルスと夾雑粒子が、どの程度の遠心時間で、どのようなショ糖密度のところに分布するか事前に確かめておく必要があります。
伊藤 できればチャンピオンデータではなく、よく起こる問題点が含まれているような具体的な実験例を示して解説していただけないでしょうか。
籔井教授 なかなか難しい要求というか、出したくないような例を出してくれとうことですね。
伊藤 すみません。しかし、その方が役立ちます。
籔井教授 それでは、1例を示しましょう。出発材料はロタウイルスの培養液1,200 mLです。これをゴミ取り遠心の後、超遠心によるペレッティングを行い、30 mLに懸濁しました。本来であれば、「10分の一の原則」により120 mLにすべきだったはずです。さて、ロタウイルスの懸濁液30 mLを6本に分け1本あたり5 mLとし、3 mLの60%(w/v)と2.5 mLの40%(w/v)のショ糖層の上に重層します。これをスウィングロータ SW 41 Tiで、38,000 rpm、4 時間遠心した後、0.5 mLずつ21本に分画して採取しました。その各フラクションについて、280 nmと260 nmでの吸光度(A280とA260)、ロタウイルス抗原検出のELISAおよびショ糖の屈折率からショ糖濃度を求めて、グラフを描きました(図)。
早斐 2つのバンドに対応して2つのピークが見られます。グラフから読み取ると、Band Aはショ糖濃度が50%のところに来ていて、Band Bは40%ですね。ただ、どちらも赤い点線で示されたA260がピークを作っているので、核酸がある。つまりロタウイルスが存在するように思いますが。
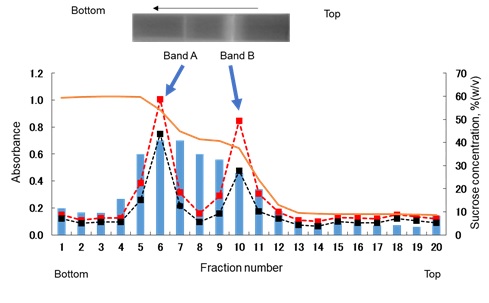
籔井教授 目で見るとBand Aの方がBand Bよりも薄いけれども、吸光度は大きい。つまり、より精製されたロタウイルス粒子がここにあるのでしょう。この付近のフラクションのA260 /A280 をみると1.3 ~1.4 くらいになっていますが、これが精製された純度の高いロタウイルス粒子のA260 /A280 比の値です。ロタウイルス粒子としては、Band BもBand Aも似たようなものが存在していると思います。しかし、Band Bは黒い点線で示したA280 の吸光度がBand Aに比べてずっと小さい割には、目で見たバンドが濃い。ですから、タンパク質以外の脂質が存在する膜成分などが絡みこんでいるのではないかと想像されます。
早斐 大きな塊になれば、S値も大きくなって早く沈むのではないですか?
籔井教授 ベックマン・コールターの遠心機カタログ223ページにある式をもう一度見てください。

注目してほしいのは(ρp―ρl)の項。つまりウイルス粒子の密度から溶媒(ウイルスを懸濁しているバッファー) の密度を引いた密度の差です。実は密度との関係は質量%(重量%)の方が簡単な対応表があるのですが、われわれは通常実験で容量%を使っています。ショ糖の容量%で60%(w/v)の溶液は、質量%では49%(w/w)に相当し、そのショ糖溶液の密度は1.29 g/cm3(mL)です。これは、ほぼ単純タンパク質の密度に相当します。また容量%で40%(w/v)の溶液は、質量%では35%(w/w)に相当し、密度は1.15 /cm3(mL)です。ロタウイルス粒子の密度は1.36 g/cm3(mL)ですが、脂質が加わるとずっと軽くなります。
早斐 コレステロール含量の多いLDL(低密度リポタンパク質)の密度は、1.019 - 1.063 g/mLだそうです。だから脂質を含んだ細胞成分がからまると粒子と溶媒の密度差は0に近くなってしまい、なかなか沈降しない。あるいは、ショ糖のクッションを貫通できなくなってしまうのですね。
籔井教授 ちなみにLDLの直径は22 nmくらいですが、これは、ちょうどB型肝炎ウイルスの表面抗原(HBs抗原)からなる小型球形粒子と同じ大きさです。小型球形粒子の密度は1.22 g/cm3(mL)です。小型球形粒子も脂質を含んでいるからです。
伊藤 話を最初に戻して、Band AとBand BのA260の比較をすると、遠心前のサンプルに存在していたロタウイルス粒子の半分近くが失われることになってしまいますね。それで前段階のペレッティングの後、濃すぎるペレット懸濁液を作らないこと、また十分に細かな懸濁液を作ることが重要なのですね。
籔井教授 そのとおりです。Band Bの中に紛れ込むロタウイルス粒子を限りなく少なくしたいのです。そして、そのためには、その前段階の分画遠心でのペレットの懸濁の仕方がカギを握っているのです。
ところで、棒グラフで示しているELISAの結果を見てください。ピークは1つで、だらだらとすそ野が広がっています。たとえショ糖クッション遠心であっても、これではオーバーロードでしょう。
伊藤 第2話の最後に下図をお示ししてお聞きした質問ですが、Band Bに相当する「不純物の層」の中にも立派なロタウイルス粒子が存在しているわけですね。それは弊社のインタビュー記事のためにデモンストレーション用の粗精製ウイルス(Band Aに相当)が必要で、収量をあまり気にする必要がなかったからというわけですね。今日のお話で、なるほど、と納得しました。

関連ページ
|
|
超遠心機 ベックマン・コールターは1947年に初めて超遠心機(超遠心分離機)を販売開始し、現在では高い遠心力・回転数はもちろん、バイオセーフティモデルやリモートコントロール機能などを備えた超遠心機を取り揃えております。様々なチューブや容量に対応するロータやアクセサリ類も取り揃え、ウイルスの高純度精製やエクソソーム分離、その他幅広いアプリケーションのご要望にお応えします。 |

