X線結晶構造解析のための膜タンパク質精製の基礎
私たちの使用している薬の50%以上が膜タンパク質と相互作用をすることが知られています。このため、薬の開発・改良には膜タンパク質を原子レベルで調べることが重要となります。ERATO“岩田ヒト膜受容体構造プロジェクト”(科学技術振興機構, 2005-2011年)に始まった一連の研究成果により、X線結晶構造解析を用いて膜タンパク質を原子レベルで理解することができるようになっています。この過程において、ボトルネックの1つであった「膜タンパク質を大量に精製する」技術が確立されました。そこで、開発者の京都大学 教授 岩田想先生と特定講師 浅田秀基先生に「X線結晶構造解析のための膜タンパク質精製の基礎」についてお話を伺いました。
(インタビュア:マーケティング本部 伊藤俊行)

岩田 想 先生

浅田 秀基 先生
先生のご研究の背景について
岩田先生 私の研究室では、膜タンパク質のX線結晶構造解析により、膜タンパク質の形状を原子レベルの分解能で調べています。つまり、その1 個1 個の原子の位置を調べるという研究を行っております。現在市場にある薬の50%以上が膜タンパク質と相互作用することが知られています。それらに作用する薬つまり低分子化合物は、今までは基本的にトライ&エラーにより開発されてきました。つまり、化学合成により膨大な数の候補化合物を合成し、その中から作用があるものを選択してきました。しかしながら、実際には、膜タンパク質に作用する部位(オルソステリック部位)が存在しますので、原子レベルでその部位の形が分かっていれば、もっと効率的に薬を作ったり、旧来あったものを改良したりすることができるようになります。このように、膜タンパク質の構造解析を通して、創薬に貢献することを目的としています。
ナノテクノロジーという言葉がありますが、その言葉が出たとき、私はすごく不満でした。なぜならば、私たちはオングストロームテクノロジー、つまりナノの10分の1の分解能で研究をしているためです。このオングストローム(Å)という領域までおりてくると、原子の位置が分かります。おおよそ原子と原子の結合距離が1から2Åの間ですので、それだけの分解能があれば、原子の位置関係を明確に知ることができます。この技術をもって膜タンパク質のオルソステリック部位を明らかにしています。この結果、効果がある低分子化合物を比較的簡単に作成できるようになってきています。ただし、その化合物がヒトに入った後の動態については今もってなお詳細は不明なことが多いのが現状であり、治験などはまだ時間がかかってしまいます。最初は、比較的発現・精製しやすい膜タンパク質をターゲットとしていましたが、最近は創薬のターゲットになりうる受容体であるヒスタミンH1受容体(Shimamura T et al.,Nature 475: 65-72, 201 1)、アンジオテンシンⅡ受容体(Asada H et al., Nat.Struct.Mol.Biol. 25(7):570-576, 2018)やプロスタグランジン受容体(Morimoto K et al., Nat Chem Biol.15: 8-10, 2019)などの構造解析を狙ってできるようになりました。この研究の過程で得られた膜タンパク質の精製に関する知見についてご紹介したいと思います。

国立大学法人 京都大学大学院医学研究科・医学部 分子細胞情報学分野 研究室のみなさま
膜タンパク質の精製について
岩田先生 X線結晶構造解析のための膜タンパク質の精製は、まず、細胞に膜タンパク質を大量に発現させ、そこから細胞膜を分離して、膜タンパク質を精製し、その結晶を得るという流れになります(Fig.1)。最終的には、目的の膜タンパク質を数ミリグラムから数十ミリグラムを取ることになります。
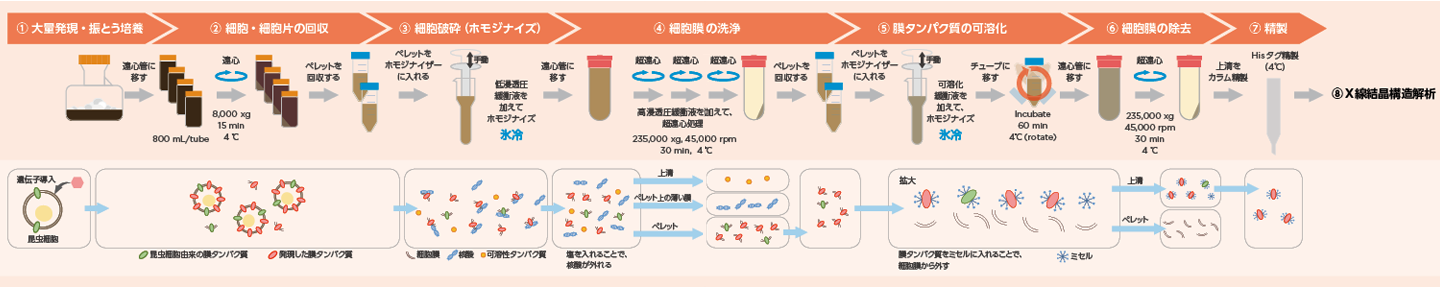
Fig.1 X線結晶構造解析のための膜タンパク質精製の概要
① 膜タンパク質の大量発現・振とう培養
岩田先生 ヒトの膜タンパク質をターゲットにしていますが、ヒト由来の培養細胞を使用しているわけではありません。私たちは酵母も使っていますが、現在は主に昆虫細胞にターゲットのタンパク質を作らせています。普通のタンパク質(膜タンパク質ではない)を遺伝子組み換えで作製する場合は、大腸菌を使うことが多いです。しかし、大腸菌は原核生物で、原核生物と真核生物では膜タンパク質の合成機構が異なり、真核生物の膜タンパク質を大腸菌で発現できる可能性はとても低いです。このため、大腸菌を用いることはできません。それならば真核生物である酵母での発現も考えられますが、酵母の場合必ずしも活性を持った形で膜タンパク質を発現させることができません。昆虫細胞の方が活性を持った形で発現されやすいことが分かっています。また、ヒト細胞は浮遊させて大量に細菌のように攪拌して培養することが難しいため、タンパク質を数ミリグラムから数十ミリグラムを取るには向きません。しかし、昆虫細胞は大量スケールでの浮遊培養を行うことができます。このため昆虫細胞を主に用いています。
浅田先生 最初の段階として、ウイルスを用いて昆虫細胞に目的膜タンパク質の遺伝子を導入して培養します。5 Lのフラスコに2.5 Lの培養液を入れて、それを複数本同時に振とう機で攪拌しながら培養します(Fig.2)。1サンプルあたり5から10 Lのスケールで培養し、目的膜タンパク質が培養液1Lあたり1 mg精製できることを目標に行っています。このときの細胞数は1.5~2.0x106 cells/mLで感染を始めて、3日間ほど培養した後に回収します。細胞数はその間、ほとんど増減はしません。

Fig.2 5 Lフラスコによる2.5 Lの培養液での大量培養(Step ①)
岩田先生 昆虫細胞の場合は、ウイルスを感染させているので、破裂しながらタンパク質を作っています。
浅田先生 死にながら発現しているのですが、あまり死にすぎるとタンパク質に悪影響を及ぼす可能性がありますので、細胞の生存率にはかなり気を使っており、8割は切らないようにして細胞を回収しています。
② 細胞・細胞片の回収
浅田先生 培養終了後、培養液5 Lの遠心処理を行い100 mLくらいの細胞のペレットにします(Fig.3)。目的の膜タンパク質は膜画分にあるので、細胞を回収すればタンパク質も回収することができます。このステップでの細胞の生死は関係ありませんので、大容量遠心機を用いて8,000 xgで15分間、4℃で遠心処理を行います。上清を捨て、得られたペレットをシリコン製スプーン(料理用)を使って、50 mLコニカルチューブに回収します(Fig.4)。この際に、PBS(-) with 20%グリセロールでボトルからできる限り細胞を回収します。次のステップまで時間が空く場合には、液体窒素で凍結し、‐80℃で凍結保存ができます。

Fig.3 遠心処理後の細胞のペレットの様子(Step ②)

Fig.4 細胞ペレットの回収(Step ②) PBS(-) with 20%グリセロールでボトルからできる限り細胞を回収する。次のステップまで時間が空く場合は、液体窒素にて凍結後、-80℃で保存する。
③ 細胞破砕(ホモジナイズ)
岩田先生 次に細胞画分に低浸透圧緩衝液(hypotonic buffer)を加えて、完全にすべての細胞を破裂させて、氷冷下でホモジナイズします(Fig.5)。タンパク質にやさしいという理由で、基本的にガラス製の手動で行うホモジナイザーを用いています(Fig.6)。機械で破砕するタイプは圧力をかけて噴き出して破砕するため、熱が発生してしまい、タンパク質へダメージを与えてしまいます。ソニケーションも同様です。
Buffers
- Hypotonic buffer(低浸透圧緩衝液): 10 mM Hepes(pH 7.5), 20 mM KCl, 10 mM MgCl2, protease inhibitor cocktail
- High osmotic buffer (高浸透圧緩衝液): 10 mM Hepes(pH 7.5), 20 mM KCl, 10 mM MgCl2, 1.0 M NaCl, protease inhibitor cocktail
- 細胞にHypotonic bufferを加え、ホモジナイズ
- 遠心(1回目)、235,000 xg(45,000 rpm)for 30 min at 4℃
- ペレットにHigh osmotic bufferを加え、ホモジナイズ
- 超遠心(2回目)、 235,000 xg(45,000 rpm)for 30 min at 4℃
- ペレットにHigh osmotic bufferを加え、ホモジナイズ
- 超遠心(3回目)、 235,000 xg(45,000 rpm)for 30 min at 4℃
- ペレットにHigh osmotic buffer with 40% glycerolでホモジナイズ( 膜画分)
- 次のステップまで時間が空く場合は、液体窒素にて凍結後、-80℃で保存
Fig.5 細胞破砕(Step③)と細胞膜の洗浄(Step④)

Fig.6 細胞破砕(Step ③)
回収した細胞をHypotonic buffer(低浸透圧緩衝液)で懸濁し、ダウンス型ホモジナイザーで潰してから超遠心用チューブへ移す。
浅田先生 昆虫細胞は、細胞壁もなく柔らかいですので、手動でも問題ありません。10から20往復出し入れし、細胞がすべて潰れるようにします。ペレットが固まっているので、最初の1回目は力が要りますが、1回通してしまえば、あとはスムーズに動きます。
④ 細胞膜の洗浄(超遠心処理)
岩田先生 ホモジナイズ後、超遠心用PCボトルアッセンブリにサンプルを移し、固定角ロータType 45 Tiを用いて(Fig.7)、超遠心処理(235,000 xg(45,000 rpm)for 30 min at 4℃)を行います。上清を破棄して、ペレットを取り出して、1.0 M NaClを加えた高浸透圧緩衝液(High osmotic buffer)で再度ホモジナイズし(※細胞破砕と同様の手法)、同条件で超遠心処理を行います。これをもう一度繰り返し行います(Fig.5)。1.0 M NaClにより細胞膜についている核酸などを外すことができます。つまり、細胞膜を洗っているわけです。
浅田先生 高浸透圧緩衝液でのホモジナイズの後、超遠心処理を行うと、細胞膜のペレットの上に粘性の高い核酸の層があるのが分かります。この層は普通にデカンテーションを行い、軽く振ると落ちてくるので、それは破棄します。また、超遠心処理を繰り返すことで、上清がきれいになってくることも確認することができます(Fig.8)。

Fig.7 岩田研究室の超遠心機

Fig.8 細胞膜の洗浄(Step ④) 左から1回目、2回目、3回目の超遠心処理後の様子上清がきれいになっていくのが分かる。
岩田先生 この上清には可溶性タンパク質などの可溶性の不純物が含まれており、このステップだけでかなりきれいに不純物を除くことができます。3回目のペレットが、目的膜タンパク質が含まれる細胞膜画分となります。タンパク質は細胞膜の中にあり、細胞膜自身が保護作用のような働きをするので、凍結することで半永久的に保存できますし、タンパク質の分解も起こらないので安心です。プロテアーゼインヒビターを入れて一連の操作を行っていますが、それでも内在性のプロテアーゼにより、少しずつ分解が進んでしまうので、この段階までは素早く一気に行うことが望ましいです。また余談ですが、この細胞膜を分画する際に、クロマトグラフィーやフィルターを用いるという選択肢もあるように思えますが、得られるサンプルのサイズの選択などの条件を厳密に決めるという観点であまり現実的ではありません。超遠心法では遠心力を合わせることで、一番望ましいところを取ることができるので、超遠心法が実験室のスケールで考えると一番優れていると思います。
⑤ 膜タンパク質の可溶化
浅田先生 膜画分が取れて次のステップが、タンパク質を精製していく最初のステップになります。膜画分に可溶化緩衝液(Solubilization buffer)を加え、ホモジナイズし(細胞破砕と同様の方法)、チューブに移し、チューブローテーターにて回転させながら4℃で60分間インキュベートします(Fig.9)。
Buffers
- Solubilization buffer(可溶化緩衝液): 50 mM Hepes(pH 7.5), 800 mM NaCl, 0.5% (w/w) DDM(dodecyl maltoside), 0.1%(w/v)CHS(cholesteryl homosuccinate), 2.0 mg/mL iodoacetamide, protease inhibitor cocktail, ligands
- 膜画分にSolubilization bufferを加え、ホモジナイズ
- incubate for 60 min at 4℃(rotate)
- 超遠心、235,000 xg(45,000 rpm)for 30 min at 4℃
- 上清のみを回収
- 精製ステップへ
Fig.9 膜タンパク質の可溶化(Step⑤)と細胞膜の除去(Step⑥)
岩田先生 この緩衝液にはDDM(dodecyl maltoside)という界面活性剤とCHS(cholesteryl homosuccinate)というコレステロールの誘導体が入っています。DDMが丸いミセル*を作り、その中に膜タンパク質が入ります。DDMのミセルが脂質二重膜から膜タンパク質を抜き取る感じです。コレステロールは膜タンパク質の安定性にとても重要で、ミセルに入った膜タンパク質を安定に保ちます。膜タンパク質にコレステロールが特異的に結合するところがあるといわれており、コレステロールは膜タンパク質に結合し、全体がDDMのミセルに覆われているといったイメージになります。一方、細胞膜は基本的にその二重膜だけになります。また、可溶化緩衝液にはリガンドを入れています。タンパク質はリガンドと結合することで形が安定し、プロテアーゼによる切断を受けにくくなります。精製前はプロテアーゼが混入しており、特に可溶化した後はプロテアーゼによる分解を非常に受けやすくなります。このため、可溶化の後は時間との勝負となります。
*ミセル:界面活性剤などの分子が水中で自己集合し、その分子が数十から数百個集まったコロイド粒子を作る。これ粒子をミセルという。⑥ 細胞膜の除去(超遠心処理)
岩田先生 インキュベート後、超遠心用PCボトルアッセンブリにサンプルを移し、固定角ロータType 45 Tiを用いて、超遠心処理(235,000 xg(45,000 rpm)for 30 min at 4℃)を行います(Fig. 9)。超遠心処理を行うことで、脂質二重膜は沈降しペレットとなり、膜タンパク質はミセルとなり可溶化しているため上清に残りますので、上清を分取します。
実は、脂質二重膜は界面活性剤の影響で変質し、半分可溶化しているようなドロンとしているような状態となっています。このため、普通の遠心機では完全に沈降しません。不純物をできるだけ除くことで精製が楽になりますので、このステップでは超遠心処理がすごく大事になります。また、可溶化の後は、プロテアーゼによる分解を最小限にするため時間との勝負となるため、短い時間で高い回転数で遠心処理を行うことが重要です。膜画分を集める段階は普通の遠心機でも長時間の遠心処理によりできなくはありませんが、このステップは超遠心一択となります。また、このステップをカラムクロマトグラフィー等の方法で行うことも考えられますが、一般的に担体であるビーズの疎水性が高く、タンパク質が非特異に吸着してしまうため、回収率が決して高くはありません。しかし、超遠心は吸着が起こりづらいので、回収率が非常に高いです。
⑦ 膜タンパク質の精製
岩田先生 上清の膜タンパク質画分からHisタグ*で目的の膜タンパク質を分取して、次にHisタグを切ってからもう一度精製すると、Hisタグはカラムに残って、目的の膜タンパク質だけを精製することができます。実は、このHisタグを切るタイミングで糖鎖修飾も切ってしまいます。結晶を作る際にタンパク質の表面同士が接触するのですが、そこに糖鎖があると、糖鎖は形が軟らかいので結晶になりにくくなります。オルソステリック部位(薬が結合するところ)はタンパク質の中にありますので、この形を決めるという目的では糖鎖は関係してきません。目的の膜タンパク質の遺伝子に変異を入れて糖鎖修飾がかからない形にすることもできますが、糖鎖修飾がたくさんある場合はすべてに変異を入れてしまうと発現が下がってしまうため、多くの場合は後から糖鎖修飾を切るようにします。Hisタグと糖鎖修飾を除いた後、つぎに会合体・凝集体を除く目的でゲルろ過を行い、できるだけ単一の状態の膜タンパク質にします。分子上決まった形だけにすることで結晶化しやすくなるためです。しかし、ゲルろ過担体のビーズは疎水性が高いため、非特異的に膜タンパク質が吸着してしまいサンプル量が減ってしまいます。よって、精製度と収量とのさじ加減が必要となります。
膜タンパク質の精製度は、SDS-PAGEで確認します。クマシー染色できちんと染まり、単一バンドであることが重要です。普通に染めて、きちんと単一バンドになっている精製度でないと結晶化のサンプルに使えません。悪いサンプルは、何本もバンドが出たりします。こういった場合は、安定性が十分でないためにプロテアーゼにより分解されてしまっていたり、糖鎖修飾の外し方が不十分であったりといったために起こります。
*Hisタグ:ヒスチジンが6個程度連続して連なっているタグペプチドで、固定化金属イオンアフィニティークロマトグラフィーにより精製することができます。⑧ X線結晶構造解析
岩田先生 X線結晶構造解析のステップは、精製した膜タンパク質を20 mg/mL以上まで濃縮して、脂質キュービック相に入れて、この脂質キュービック相の中で結晶が成長するのを待ちます。脂質の種類と条件によって三次元の連続につながった相を作ることがあって、それが脂質キュービック相です。ここに、膜タンパク質を入れます。膜タンパク質は、最初は溶液中では界面活性剤のミセルの中に溶けています。ところが、キュービック相の中に入ると、基本的にはキュービック相自身の脂質が界面活性剤を吸収してしまって、膜タンパク質はキュービック相の中に入り、自由に動くこと(拡散すること)ができるような状態になります。この状態で結晶化する方法が非常に有効であることが分かりました。実際の手順をお話しします。小さなウェル(くぼみ)のあるガラスプレートに(Fig. 10)、脂質キュービック相とタンパク質を混ぜたものを置いて、その上から沈殿剤の溶液をかけます。ガラスプレートでフタをし、結晶が成長するのを待ちます。数日から1 週間くらいまでで結晶化すると良い結果が得られることが多いです。

Fig.10 結晶化に用いるプレート
それ以上の日数では、タンパク質が分解する可能性が高まるため、上手くいくことが少ない印象です。逆に混ぜてすぐや一晩といった極端に短い時間で結晶化する場合は、格子欠陥といって、きれいに分子が積み上がっていない状態であり、構造解析には使えません。キュービック相の中で結晶を作ると、顕微鏡でないと見えないくらいの小さな結晶しか取れません。結晶は最初に核ができて、そこにタンパク質がついて結晶ができていきます。しかし、キュービック相自身が非常に粘調なので、タンパク質の拡散が緩慢なため、一つの大きな結晶になりにくく、小さな結晶が沢山できる傾向にあります。
昔はこの小さな結晶からX線結晶構造解析を行うことは難しかったのですが、今は幸いなことに非常に細い数μmくらいのX線ビームを使ってデータを取ることができるようになりましたので、小さな結晶でも構造解析ができます。小さな結晶が複数個できますので、一個一個拾うのでなく、それを丸ごとX線の装置に載せて、自動でX線を端から照射していきます。結晶が目に見えないので、反射があることでそこに結晶があったことが分かります。この結晶があった場所からデータを取って構造解析を行います。このX線結晶構造解析はSPring-8で行い、また、SACLAにあるX線自由電子レーザーを使っての構造解析も行っています。
ご研究の展望について
岩田先生 私自身が今すごく興味を持っているのは、SACLAのX線自由電子レーザーを使ってのタンパク質の時間分割構造解析です。SACLAを使うと、例えば酵素反応中の原子の動きをスナップショットにして撮ることができます。普通のシンクロトロン放射でデータを取る際は、結晶に数分の1秒から1秒間X線を当てて、その時の回折データを取得します。一方、SACLAは、10フェムト秒(10 x 10-15秒)というパルスです。つまり、1回のイメージを撮るのに、10フェムト秒でデータ取得ができます。暗室でストロボライトを使って写真を撮るのと同じで、タンパク質が動いている時に光(レーザー)を当てると、その瞬間が止まったタンパク質の形を撮ることができます。ただ、少し欠点があって、ストロボライトは当たっても壊れませんが、X線自由電子レーザーの場合は、一回の照射でタンパク質がプラズマになってしまいます。このため、異なる結晶に対して時間を変えながら何回か同じ実験を行い、データを組み合わせることで、酵素の反応中の動きを調べます。この技術を使って光がスイッチとなって活性・非活性が切り替わるタンパク質の仕組みを解明したいと思っています。このようなタンパク質は、今は基本的にはイオンチャネルを形成しています。つまり、光が当たるとチャネルが開くとか、閉じるとかを行っています。さらに、光によるスイッチのメカニズムが分かってきたら、そういったタンパク質の光とかで制御できるリガンドになる化合物が合成できます。それを用いて、膜タンパク質の機能を制御できるようにしたいと思っています。
最後にベックマン・コールターについて一言
岩田先生 私は、自分の研究生活を通して、ベックマン・コールター以外の超遠心機を使ったことがありません。超遠心機は少し間違うと、簡単にサンプルが駄目になります。大事なサンプルの処理をしますので、やっぱり信頼をおけるのが一番だと思います。私の研究室は、超遠心機が5台並んでおり、毎日使用していますので、サービス体制も重要です。安定して超遠心機を5台稼働することができているのは、ベックマン・コールターの信頼性と充実したサービス体制のおかげだと思います。

施設紹介

国立大学法人 京都大学大学院
医学研究科・医学部 分子細胞情報学分野
生体の恒常性維持や病態発生といった医学的にも重要なさまざまな生命現象の素過程は、微視的には生体分子の「かたち」や分子間相互作用という物理量を基盤として成り立っています。当研究室では,種々のヒト疾患の発症機構にかかわり、かつ多くの医薬品の作用点となっている膜タンパク質やその複合体を主な対象として、X線結晶構造解析を進めています。また、計算機を用いた新規医薬品の合理的な分子設計・探索、分子構造の動きのシミュレーションなどにも意欲的に取り組み、物質構造科学の立場から細胞機能制御の原理を探究しています。
